
Häufig gestellte Fragen zum Batchsystem
Jobs vorbereiten
Nur mit diesen Pflichtangaben kann der Scheduler entscheiden, auf welchen Compute-Knoten er den Benutzerjob zur Ausführung bringt.
Wenn man z.B. kein --mem-per-cpu= angibt, würde ein Job mit sehr großen Hautspeicheranforderungen u.U. auf einem Knoten mit zu wenig RAM ausgeführt werden und abstürzen.
Um es „spielerisch“ auszudrücken: mit den Ressourcenanforderungen aller im Batchsystem eingestellten Jobs muss der Scheduler eine Art „multidimensionales Tetris“ spielen, um die Jobs mindestens nach den Dimensionen Laufzeit, Hauptspeicherbedarf und CPU-Core-Anzahl möglichst effizient im Cluster zu plazieren. (Im Hintergrund spielen noch viele andere Parameter als diese drei eine Rolle.)
Mindestens diese drei muss man also angeben, um dem Scheduler etwas zum Verteilen an die Hand zu geben.
Ja, das geht mit dem Parameter „--test-only“ bei „sbatch“:
sbatch --test-only submit_tests
[E] sbatch: Job 12…78 to start at yyyy-mm-ddThh:mm:ss using XX processors on nodes mpsc[000y-000z] in partition PAntwortet das System mit solch einer Meldung, würde Slurm diesen Job so annehmen und voraussichtlich zu der (geschätzten!) Zeit anlaufen lassen.
Obwohl das System auch diese Meldung scheinbar als Fehler klassifiziert („[E] …“), ist bei obiger Ausgabe kein echter Fehler aufgetreten.
Sollten Sie aber z.B. einen erforderlichen Parameter vergessen haben, sähe die Ausgabe zum Beispiel in etwa so aus:
[E] Missing runlimit specification – Use --time=<[hh:]mm>Wenn sich sbatch <jobscript> über fehlende Angaben beschwert, obwohl Sie genau diese in Form von Pragmas
#SBATCH …
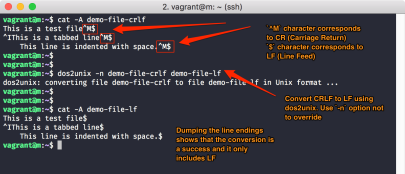
definiert haben, kann das an Windows-Zeilenenden liegen, die von UNIX/Linux nicht als gültig erkannt werden.
Wenn Sie das fragliche Jobscript auf Ihrem Windows-PC erzeugt und per scp auf einen Loginnode übertragen haben, wandeln Sie es einfach per
dos2unix jobscriptfilein ein UNIX/Linux-File um.
Sollten die obigen Fehler dann immer noch auftreten, überprüfen Sie die Minus- bzw. Bindestrich-Zeichen „-“. Hier können sich Gedanken- oder Geviertstriche eingeschlichen haben, die nicht als Parameter-Einleitungszeichen wirken können.
Bevor Sie Jobs abschicken, müssen Sie ermitteln, wieviele CPUs (= Kerne) Sie am besten nutzen, wieviel Hauptspeicher Ihr wissenschaftliches Programm benötigen wird und wie lange es für die Berechnungen mindestens laufen muss.
Falls Ihr wissenschaftliches Programm und vergleichbare Probleme in Ihrer Arbeitsgruppe bereits bearbeitet werden, erkundigen Sie sich bei Ihren Kollegen nach deren Erfahrungen damit.
Falls Sie mit neuem Programm oder neuem Problem starten: bereiten Sie sich einen vergleichsweise kleinen Testfall vor (nicht mehr als 30 Minuten Laufzeit) und lassen Sie ihn auf einem der Login-Knoten mit der gewünschten Anzahl CPU-Kernen unter der Kontrolle des UNIX-„time“-Kommandos laufen:
/bin/time --format='MaxMem: %Mkb, WCT: %E' myProgram <testcase>Danach erhalten Sie auf dem STDERR-Kanal zum Beispiel die Ausgabe
- MaxMem: 942080kb, WCT: 1:16.00
Nach Umrechnung von „MaxMem“ in MBytes (durch 1024 dividieren) können Sie Ihren #SBATCH --mem-per-cpu=-Parameter für diesen Testlauf wie folgt errechnen:
MaxMem in MByte
--------------------- (zuzüglich Sicherheitspuffer)
# der Prozessorkerne Ihre maximal benötigte Laufzeit ( #SBATCH -t d-hh:mm:ss im Jobscript) ist dann die obige WCT" (wiederum zzgl. Sicherheitszuschlag).
In unserem Beispiel und falls Sie 4 Prozessorkerne benutzt haben:
942080 / 1024 / 4 =
--mem-per-cpu=230Wenn Sie Ihren Testfall mit jeweils 2, 4, 8 und 16 Prozessorkernen laufen lassen, bekommen Sie einen groben Eindruck von der Skalierbarkeit Ihres Problems, und können die für Ihre echten Jobs nötige Laufzeit und den Hauptspeicherbedarf besser abschätzen.
In Kürze und hierarchisch dargestellt: der Lichtenberg-Cluster besteht aus
- Rechenknoten
einzelne, unabhängige Rechner vergleichbar einem PC/Laptop (nur mit mehr und leistungsfähigerer Hardware)
Ein Rechenknoten besteht aus- zwei oder mehr CPUs (central processing units oder Prozessoren), die je in einem eigenen „socket“ stecken.
CPUs sind die eigentlichen „programmausführenden“ Komponenten eines Knotens.
Eine CPU besteht aus- mehreren Kernen, die man als separate Ausführungseinheiten innerhalb einer CPU verstehen kann.
Je mehr Kerne, desto mehr unabhängige Prozesse bzw. Ausführungsstränge (threads) können gleichzeitig ausgeführt werden.
Jeder Kern kann entweder von- einem Prozess bzw. Task (MPI)
oder - einem Ausführungsstrang (thread) benutzt werden
Solch „Multithreading“ kann z.B. mit POSIX-Threads oder heutzutage meistens OpenMP (Open MultiProcessing) umgesetzt sein.
- einem Prozess bzw. Task (MPI)
- mehreren Kernen, die man als separate Ausführungseinheiten innerhalb einer CPU verstehen kann.
- zwei oder mehr CPUs (central processing units oder Prozessoren), die je in einem eigenen „socket“ stecken.
Eine reine MPI-Applikation würde also so viele verschiedene Prozesse (= Tasks/MPI-Ranks) starten wie insgesamt an Rechenkernen (cores) angefordert wurden. Diese Prozesse kommunizieren dann miteinander über MPI.
Die Prozesse eines MPI-Jobs können auch über mehrere Rechenknoten verteilt laufen, wobei die MPI-Transfers dann über den Interconnect (Infiniband) gehen.
Eine reine Multithreaded-Applikation hingegen startet nur einen Prozess, der sich dann selbst in mehrere unabhängige Ausführungsstränge aufspaltet. Jeder dieser Threads hat im Optimalfall einen einzelnen Kern zur Verfügung, so dass ein einziger Prozess viele Kerne gleichzeitig nutzen kann.
Die heute üblichen Programme nutzen dazu meist OpenMP (siehe $OMP_NUM_THREADS in der Dokumentation Ihres Programms).
Solche Applikationen können nicht über mehrere Knoten verteilt rechnen, dafür aber alle Kerne eines Rechenknotens im selben Programm/Prozess nutzen.
Hybride Applikationen kombinieren beide Parallelisierungsverfahren: pro verfügbarem Rechenknoten starten sie z.B. nur einen Prozess (= Task), der dann wiederum die auf jedem einzelnen Knoten verfügbaren Kerne mittels Threads ausnutzt. Die Threads kommunizieren verzögerungsfrei über ihren gemeinsamen Hauptspeicher innerhalb eines Knotens, während die Prozesse wiederum mittels MPI über den Interconnect (Infiniband) die Knotengrenze überwinden.
Aus historischen Gründen (und der Ära vor den heutigen Mehrkern-CPUs) hat SLURM Parameter, die scheinbar auf CPUs zugeschnitten sind (z.B. --mem-per-cpu=).
Heutzutage meint das jedoch immer Kerne statt CPUs! Das mag verwirrend sein, aber die Regel ist einfach: benutzen und berechnen Sie Ihre „--mem-per-cpu“-Werte einfach so, als hieße der Parameter „--mem-per-core“.
Für sehr viele ähnliche Jobs raten wir dringend davon ab, Shellscript-Schleifen um „sbatch / squeue“ herum zu basteln. Nutzen Sie für >20 gleichartige Jobs stattdessen einfach Job Arrays in Slurm.
Dieses Vorgehen entlastet nicht nur den Slurm-Scheduler bei seiner Arbeit, sondern Sie können auch wesentlich mehr Array-Tasks submittieren als Einzeljobs!
Beispiele für Anwendungsfälle sind:
- dasselbe Programm, dieselben Parameter, aber jeweils verschiedene Input-Daten
- dasselbe Programm, dieselben Input-Daten, aber jeweils verschiedene Parameter-Sets
- nicht-parallelisiertes Programm (das also nicht mehrere Cores [Multi-Threading] oder gar mehrere Nodes [MPI] nutzen kann) – dasmit dem Sie aber viele separate Input-Daten bearbeiten wollen, wobei kein Einzellauf von Ergebnissen eines anderen abhängt (z.B. Einzelbildanalyse), auch als High-Throughput Computing bezeichnet
Benennen Sie die jeweils „zahlreichen“ Bestandteile mit durchgehender Numerierung: image1.png, image2.png oder paramSet1.conf, paramSet2.conf etc.
Haben Sie z.B. 3124 Sets, setzen Sie ein Jobscript mit
_______________
#SBATCH -a 1-3124
myProgram image$SLURM_ARRAY_TASK_ID.png > image$SLURM_ARRAY_TASK_ID.png.out_______________
auf und submittieren Sie es. Slurm startet jetzt einen einzigen Job mit 3124 ArrayTasks, die jeweils ihre eigene Bilddatei analysieren und das Ergebnis in jeweils ihre eigene Ausgabedatei schreiben.
Auch für die mittels „-o“ und „-e“ spezifizierten Ausgabe- und Fehlerdateien können Sie „%.-Platzhalter nutzen:
#SBATCH -o /path/to/my/dir/out_%A_%a.txt
#SBATCH -e /path/to/my/dir/err_%A_%a.txt %A = $SLURM_ARRAY_JOB_ID (die “übergordnete„, gemeinsame Job-ID – identisch für alle ArrayTasks)
%a = $SLURM_ARRAY_TASK_ID (die unterschiedliche, hochzählende Nummer jeder einzelnen ArrayTask)
Wenn Sie die Anzahl gleichzeitig laufender ArrayTasks limitieren müssen, nutzen Sie
#SBATCH -a 1-3124%10
Dann lässt Slurm höchstens 10 Tasks gleichzeitig laufen.
Weitere Details finden Sie in 'man sbatch' unter “--array="., oder auf der Slurm-Website .
Falls Ihr Programm über „Checkpoint/Restart“ (CPR) verfügt, können Sie auch dieses Problem mit einem Job Array lösen, allerdings mit einem seriellen.
Weisen Sie Ihr Programm an, seine Zwischenstände der Berechnungen/Simulation regelmäßig in ein „state file“ auf /work/scratch/…/ zu schreiben.
Einerseits schützt das Ihre Jobs vor Hardware-Ausfällen, z.B. wenn einer der beteiligten Rechenknoten abstürzt.
Ihr Programm muss dann nicht von vorn beginnen, sondern kann den Zwischenstand aus dem letzten „state file“ lesen und von da aus weiterrechnen.
Andererseits können Sie damit auch ein „serielles“ Job Array aufsetzen, dass aus einer Folge von 24h-Teiljobs besteht. Jeder Job wird zwar nach 24h abgebrochen (TIMED OUT) – aber das serielle Array startet sofort den nächsten 24h-Job, der das „state file“ seines Vorgängers einliest und für die nächsten 24h daran weiterrechnet.
Mit dieser Methode können Sie weit längere Simulationen als 7 Tage ausführen.
Angenommen, Ihre Simulation benötigt 19 Tage Laufzeit – dann würde das folgende Array 19 einzelne, nacheinander ablaufende Jobs mit jeweils 1 Tag Laufzeit erzeugen:
_______________
#SBATCH -a 1-19%1
#SBATCH -t 1-
srun mySimulationProg …_______________
Der Parameter %1 (direkt hinter die Array-Indizes geschrieben) sagt dem Array, zeitgleich immer nur einen der Array-Jobs laufen zu lassen, und danach den nächsten.
Nach dem Submittieren startet Slurm irgendwann den ersten Job, bricht ihn nach 24h ab und läßt den zweiten folgen – und so weiter bis zum 19. Tag.
Weitere Details finden Sie in 'man sbatch' unter „--array=“., oder auf der Slurm-Website .
Den meisten wissenschaftlichen Programmen kann man die Zahl der zu nutzenden CPU-Kerne bzw. GPUs als Parameter mitteilen.
Anstatt nun jedesmal sowohl die „#SBATCH …“-Anforderungszeilen als auch die Kommandozeile Ihres Programms ändern zu müssen, können Sie Slurm-Variablen nutzen. Diese werden zum Zeitpunkt der Job-Allokation (Start) gesetzt und stehen im Script zur Verfügung.
Wenn Ihr Programm zum Beispiel MultiThreading in Form von OpenMP nutzt, können Sie Ihr Job-Skript folgendermassen vereinfachen (# = Anzahl):
_______________
#!/bin/bash…#SBATCH -c ##export OMP_NUM_THREADS=$SLURM_CPUS_PER_TASK/Pfad/zu/meinem/wissenschaftlichenProgramm …_______________
Es wird dann immer genau die Anzahl an CPU-Kernen nutzen, die Sie mit „-c ##“ angefordert haben.
Gleiches gilt für GPUs. Wenn die Dokumentation Ihres Programms erklärt, dass die Anzahl der zu nutzenden GPU-Karten beispielsweise mit dem Parameter „--num-devices=#“ anzugeben ist (# = Anzahl):
_______________
#!/bin/bash…#SBATCH --gres=#/Pfad/zu/meinem/wissenschaftlichenProgramm --num-devices=$SLURM_GPUS_ON_NODE … _______________
Auf diese Weise müssen Sie nur die #SBATCH-Zeilen mit „-c ##“ bzw. „--gres=#“ anpassen, und Ihre Programme laufen automatisch immer mit den tatsächlich angeforderten Ressourcen.
(Bitte übernehmen Sie das # nicht direkt in Ihre Scripts – ersetzen Sie es durch die gewünschte Zahl der Kerne/GPUs.)
Hier können Sie die „--multi-prog“-Option von srun nutzen – damit werden in einer einzigen Allocation (=Job) verschiedene Programme gestartet.
#!/bin/bash #SBATCH … srun … --multi-prog myCtrlWorker.conf_______________Die Datei myCtrlWorker.conf ist nach dem Schema
Nr /path/to/program <parameter>aufgebaut, und enthält pro Zeile die Definition, welcher MPI-Rank („Nr“ ⇦ #SBATCH --ntasks=XX) was genau mittels srun und mit welchen Parametern starten soll.
Sollten die „Worker“-Instanzen einen Parameter für ihre jeweils eigene „Nummer“ verlangen, können Sie dort die Platzhalter „%t“ bzw. „%o“ verwenden (siehe ' man srun' unter MULTIPLE PROGRAM CONFIGURATION).
Beispiel
… für einen Controller, der 24 Worker ansteuert:
_______________ # MPI rank, then path to binary 1 /path/to/controller_program <parameter for controller> 2-25 /path/to/worker_program <parameter for worker, z.B. WorkerID=%o>_______________Hier wird srun also zuerst (1) den Controller starten, danach 24 einzelne (2-25) „Worker“-Instanzen, die ihre jeweilige Nummer durch %o erfahren.
Beispiel
… für beliebig viele Worker (Anzahl nur bestimmt durch #SBATCH --ntasks=XX):
_______________ # MPI rank, then path to binary 1 /path/to/controller_program <parameter for controller> * /path/to/worker_program <parameter for worker, z.B. WorkerID=%o>_______________Der Stern * steht hier für „alle weiteren Ranks“ (ab 2 aufwärts).
Wie genau Controller- und Worker-Prozesse dann im Hauptspeicher „zueinander finden“ bzw. welche Parameter dazu nötig sind, entnehmen Sie bitte der Programm-Dokumentation.
Ja, das geht – mittels
#SBATCH --mail-type=FAIL,ENDin Ihrem Jobscript erhalten Sie Mails über fehlgeschlagene und beendete Jobs.
Wir empfehlen dabei, den Parameter
#SBATCH --mail-user=your@email.address.hereüberhaupt nicht zu benutzen, weil
- unser System die Standard-Mailadresse zu Ihrer TU-ID automatisch herausfindet
- Schreibfehler in your@email.address.here (beispielsweise mit spitzen Klammern agegeben:
<…>) immer zu Fehlleitungen oder Rückläufern („unzustellbar“) führen, und diese erzeugen bei uns nutzlose Service-Tickets - Sie sollten hier nie Ihre private EMail-Adresse angeben! Eine möglicherweise aus vielen Ihrer Jobs entstehende „Mailflut“ führt dazu, dass Ihr privater Mailanbieter unsere TUDa-Mailserver auf seine Spam-Blacklist setzt und jegliche Annahme verweigert.
Das blockiert dann nicht nur Ihre Mails, sondern alle Mails, die von irgendwelchen TUDa-Mailadressen an Ihren Mailanbieter gehen sollen – stellen Sie sich das für@gmail.comoder@web.devor…!
Slurm verweigert die Annahme Ihres Batch-Jobs mit dieser Fehlermeldung, wenn Sie ein nicht existierendes oder bereits ausgelaufenes Projekt (#SBATCH -A) angeben.
- Prüfen Sie mittels „
member“, ob Sie bereits Mitglied sind - Prüfen Sie Ihre Umgebungsvariablen, ob Sie eventuell darüber ein ausgelaufenes Projekt ansteuern:
env | grep ACCOUNT
Hier könnte z.B. „SBATCH_ACCOUNT=<veraltetes Projekt>“ auftauchen, was Ihre „#SBATCH -A …“-Zeile übersteuert
Ebenfalls könnte es sein, dass Sie zuviele Batch-Jobs abzuschicken versuchen.
Nutzen Sie statt vieler Einzeljobs besser Job Arrays (siehe weiter oben auf dieser Seite)!
Wartende Jobs
Die von Slurm-Kommandos wie „squeue“ oder „sprio“ angezeigten Prioritätswerte sind immer relativ zueinander und relativ zur momentanen Last auf dem Cluster zu verstehen. Es gibt keinen „Absolut“- bzw. „Schwellwert“ der Priorität, ab der Jobs unabdingbar anlaufen.
So kann in Phasen geringer Last auf dem Cluster bereits ein kleiner Prioritätswert genügen, um Jobs (auf den freien Ressourcen) umgehend anlaufen zu lassen. Zu Zeiten hoher Last und gar keiner freien Ressourcen kann hingegen selbst ein viel höherer Prioritätswert nicht verhindern, dass die Jobs lange warten müssen.
Da die Standardlaufzeit von Jobs 24h beträgt, müssen in Phasen hoher Last auch mindestens 12-24 Stunden Wartezeit einkalkuliert werden.
Sie können sich mit dem Kommando „squeue --start“ vom Scheduler eine Schätzung geben lassen, wann ungefähr Ihre Jobs anlaufen werden.
Die Ausgabe „N/A“ ist dabei völlig normal, denn der Scheduler betrachtet nicht bei jedem Scheduling-Durchlauf jeden Job. So kann es eine Weile dauern, bis der Scheduler auch nur zu dieser „Schätzung“ für Ihre wartenden Jobs gelangt.
Generell hängt die Wartezeit eines Jobs nicht nur von der Priorität Ihrer Jobs ab, sondern auch von der momentanen Auslastung des Clusters. Darum läßt sich ganz allgemein keine 1:1-Beziehung zwischen Priorität und Wartezeit errechnen.
Auf dem Lichtenberg-HLR läuft der Scheduler im „Fair Share“-Modus: je mehr Rechenzeit Sie verbrauchen (insbesondere über Ihr monatliches Projektkontingent hinaus), desto geringer wird Ihre Priorität.
Diese Reduktion Ihrer Priorität hat aber eine Halbwertszeit von ca. 14 Tagen, so dass sich Ihr Wert über die Zeit hinweg „erholt“.
Am besten verbrauchen Sie Ihre Rechenzeit gleichmäßig über die gesamte Projektlaufzeit (siehe 'csreport'), dann sinkt Ihre Priorität auch nur moderat ab.
Bei einer geplanten Auszeit teilen wir dem Batchsystem vorher mit, wann die Auszeit beginnt. Aufgrund Ihrer Angaben, wie lange Ihre jeweiligen Jobs laufen werden (#SBATCH -t d-hh:mm:ss im Jobscript), entscheidet der Batch-Scheduler, ob Ihr Job sicher vor der Auszeit beendet werden kann. In diesem Fall lässt er ihn noch anlaufen.
Wartende Jobs (Status „PENDING“), die nicht mehr vor der Auszeit beendet werden können, laufen also auch gar nicht mehr an.
Alle wartenden Jobs in allen Queues (Status „PENDING“) bleiben über Auszeiten und Ausfälle hinweg erhalten, und werden nach der Auszeit wie gewohnt prioritätsgesteuert anlaufen.
Laufende Jobs
Prüfen Sie das Vorhandensein von allen Verzeichnissen, die Ihr Jobscript angibt, und ob Sie dort schreibberechtigt sind.
Insbesondere das Verzeichnis, das Sie mit
#SBATCH -e /path/to/error/directory/%j.errspezifizieren, muss bereits vor Anlaufen des Jobs existieren und für Sie beschreibbar sein.
Slurm bricht Ihren Job sofort und kommentarlos ab, wenn es wegen eines fehlenden Verzeichnisses die Output- und Error-Dateien nicht schreiben kann.
Ein Konstrukt innerhalb des Jobscripts wie
#SBATCH -e /path/to/error/directory/%j.err
mkdir -p /path/to/error/directory/ist ein „Henne / Ei“-Problem und kann daher auch nicht funktionieren: für Slurm ist der „mkdir“-Befehl ja Bestandteil des Jobs, dessen eventuelle Ausgaben (STDOUT oder STDERR) in das noch nicht existierende Verzeichnis geschrieben werden müssten.
Stellen Sie sicher, dass alle relevanten Module in Ihrem Jobscript geladen werden.
Sie können die nötigen Module zwar nach dem Login direkt auf dem Loginknoten laden, weil diese dann von „sbatch myJobScript“ an den Job „vererbt“ werden, aber das ist nicht zuverlässig.
Stattdessen macht es Ihre Jobs von den in der Login-Umgebung geladenen Modulen abhängig.
Darum empfehlen wir, jedes Jobscript mit
_______________
module purge
module load <each and every relevant module>_______________
zu beginnen, um genau die (und nur die) Module zu laden, die Ihr wissenschaftliches Programm benötigt.
Das erleichtert auch die spätere Reproduzierbarkeit Ihrer Jobs, ganz unabhängig von dem Modulset, was Sie damals in Ihrer Login-Session geladen hatten.
Dieses Verhalten kann auftreten, wenn der Job mehrfache verschachtelte Aufrufe von srun (oder mpirun) enthält. Der zweite („innere“) Aufruf versucht dann, dieselben Ressourcen zu allozieren wie der erste („äußere“), und das kann nicht gehen.
1. Script statt ausführbares Programm:
Wenn in Ihrem Jobscript z.B.
srun /path/to/myScientificProgramsteht, prüfen Sie, ob „/path/to/myScientificProgram“ nicht vielleicht ein Script ist, das selbst wiederum srun (oder mpirun) aufruft.
In diesem Fall entfernen Sie einfach das srun vor /path/to/myScientificProgram.
2. Allokation per #SBATCH und per 'srun':
Eine solche unabsichtliche „Verschachtelung“ ist auch ein Jobscript mit zwei Allokationen: die „äußere“ per #SBATCH -n 16 und die „innere“ mit srun -n 16:
#SBATCH -n 16…srun -n 16 … /path/to/myScientificProgramLassen Sie hier einfach das „-n 16“ bei 'srun' weg, denn der Vorteil von srun gegenüber mpirun ist ja gerade, dass srun von Slurm alles über den Job weiß und „erbt“.
Beispiel-Fehler:
srun: Job XXX step creation temporarily disabled, retrying
srun: error: Unable to create step for job XXX: Job/step already completing or completed
srun: Job step aborted: Waiting up to 32 seconds for job step to finish.
slurmstepd: error: *** STEP XXX.0 ON hpb0560 CANCELLED AT 2020-01-08T14:53:33 DUE TO TIME LIMIT ***
slurmstepd: error: *** JOB XXX ON hpb0560 CANCELLED AT 2020-01-08T14:53:33 DUE TO TIME LIMIT ***OpenMPI hat unter SLURM die Einschränkung, dass genau diese Routine MPI_Comm_spawn() zum späteren Nachstarten von MPI-Ranks nicht funktioniert.
Wenn Ihr Programm dieses Feature unbedingt nutzen muss, bleibt nur der Wechsel zu einer anderen MPI-Implementierung wie z.B. „intelmpi“ übrig.
Beispielfehler:
[mpsc0111:1840264] *** An error occurred in MPI_Comm_spawn[mpsc0111:1840264] *** reported by process [2377252864,38][mpsc0111:1840264] *** on communicator MPI_COMM_SELF[mpsc0111:1840264] *** MPI_ERR_SPAWN: could not spawn processes[mpsc0111:1840264] *** MPI_ERRORS_ARE_FATAL (processes in this communicator will now abort,[mpsc0111:1840264] *** and potentially your MPI job)Slurm kann nicht erkennen, welches der Kommandos in einem Job (script) wirklich „wichtig“ ist. Der einzige Mechanismus zur Feststellung von Erfolg oder Fehler ist der exit code Ihres Jobscripts, nicht der „echte“ Verlauf Ihres wissenschaftlichen Programms.
Programme, die sich an gängige Programmierkonventionen halten, beenden sich mit einem exit code von 0, wenn alles geklappt hat, und >0 im Fehlerfall.
Der exit code eines Skripts ist der seines letzten Kommandos.
Im folgenden Beispiel
#!/bin/bash
#SBATCH …
myScientificProgram …ist das zuletzt ausgeführte Kommando wirklich das wissenschaftliche Programm, und damit endet das gesamte Jobscript wie gewünscht auch mit dessen exit code. Slurm wird den Job nun als COMPLETED vermelden, wenn „myScientificProgram“ mit 0 geendet hat, und als FAILED, falls nicht.
Fügt man nun aber irgendwelche anderen Kommandos (zum Aufräumen oder Resultate-Kopieren) nach „myScientificProgram“ ein, überschreiben diese den exit code des eigentlichen Programms:
_______________
#!/bin/bash
#SBATCH …
myScientificProgram …
echo „Job beendet“_______________
Jetzt bestimmt das Ergebnis des „echo“-Befehls den exit code des gesamten Jobscripts, weil es sein letztes Kommando ist. Klappt der „echo“-Befehl (und das tut er fast immer), wird Slurm den Job als COMPLETED betrachten und so vermelden, obwohl „myScientificProgram“ eine Zeile vorher komplett fehlgeschlagen sein könnte.
Um das zu vermeiden (und trotzdem nach der eigentlichen wiss. Rechnung noch Kommandos auszuführen), muss man den „wichtigen“ exit code vor den nächsten Kommandos zwischenspeichern:
_______________
#!/bin/bash
#SBATCH …
myScientificProgram …
EXITCODE=$?
/any/other/job/closure/cleanup/commands …
echo „Job beendet“
exit $EXITCODE_______________
Hier wird der exit code von „myScientificProgram“ in der Variablen $EXITCODE zwischengespeichert. Trotz der darauffolgenden Kommandos kann man das Jobscript nun via exit-Befehl mit dem „echten“ exit code beenden, der Ihnen wichtig ist.
Dadurch empfängt Slurm jetzt den exit code der wiss. Berechnung (statt den des zufällig letzten Kommandos im Jobscript) und wird nur noch solche Jobs als COMPLETED vermelden, bei denen „myScientificProgram“ fehlerfrei durchgelaufen ist.
Wenn Sie für jeden Job wissen wollen, wann genau er startet und endet, können Sie dies mit je einer einzigen Zeile in den STDERR-Kanal schreiben lassen (den die mit „#SBATCH -e …“ angegebene Datei aufzeichnet):
_______________
#SBATCH …
/usr/bin/date +„Job ${SLURM_JOBID} START: %F_%T.%3N“ >&2
module purge
module load module1 module2 module3
/path/to/my/scientific/program …
EXITCODE=$?
/usr/bin/date +„Job ${SLURM_JOBID} END: %F_%T.%3N“ >&2
exit ${EXITCODE}_______________
Ohne extra Zeilen mit „echo“ o.ä. gibt Ihnen jetzt das „date“-Kommando die exakten Zeiten aus, und Sie haben immer die genaue Laufzeit Ihrer „Payload“.
Zu den mit % spezifizierten Platzhaltern für die Datums- und Zeitangaben konsultieren Sie einfach 'man date' unter „FORMAT controls“.
Nur während der Laufzeit eines Ihrer Jobs, und nur die Rechenknoten, die Ihre(n) Job(s) gerade ausführen.
Details dazu finden Sie in unserer „SLURM-Sektion (wird in neuem Tab geöffnet)“.
Sonstiges
In unserer „Verschiedenes zu Linux“-Sektion erklären wir, wie grafische Programme und grafische Resultate am besten darzustellen sind
Wenn Sie per Mail über Statusänderungen Ihrer Jobs benachrichtigt werden wollen, schreiben Sie in Ihrem Job-Skript
#SBATCH --mail-type=…und setzen den Parameter … auf BEGIN oder END oder FAIL oder ALL, und der Scheduler wird die entsprechenden Mails an Sie versenden.
Da alle Jobs immer mit Ihrem Login-Namen – also Ihrer TU-ID – verknüpft sind, haben wir Slurm so eingestellt, dass es automatisch die zu Ihrer TU-ID gehörige eMail-Adresse verwendet. Diese wird nicht bei uns gespeichert, sondern immer aktuell aus dem IDM-System der TU abgefragt.
Ändern Sie also die Mailadresse Ihrer TU-ID, gehen die nächsten Scheduler-Mails sofort an das dort (neu) eingestellte Ziel.
Wie die Rechenknoten sind auch unsere Login-Knoten nicht mit herkömmlichen Methoden auf Festplatten installiert. Stattdessen holen sie sich bei jedem Neustart (und somit auch nach jeder Auszeit) ein Betriebssystem-Abbild über das Netzwerk und entpacken es in ihrem Hauptspeicher.
Damit haben sie nach jedem Neustart einen genau definierten (und getesteten) Zustand.
Da „cron“- und „at“-Aufträge im Systembereich gespeichert werden und dieser nach einem Neustart zurückgesetzt ist, sind Cron-Einträge nicht permanent und darum nicht verlässlich.
Damit Nutzer nicht doch welche anlegen (und sich auf deren Funktion z.B. zum Backup verlassen), haben wir „cron“ und „at“ lieber ganz abgeschaltet.
Die Zugriffsrechte in den Projekt-Verzeichnissen werden über die jeweiligen UNIX-Gruppen namens <PrjID> verwaltet.
Dateien (und Verzeichnisse), die nicht der jeweiligen UNIX-Gruppe gehören oder die nicht die korrekten Zugriffsrechte haben, sind für andere Projektmiglieder gesperrt.
Verzeichnisse und Dateien irgendwo unterhalb von /work/projects/:
- müssen der entsprechenden Gruppe
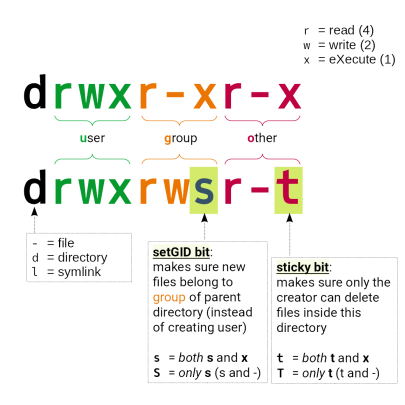
<PrjID>gehören (und nicht Ihrer TUID-Gruppe) - Verzeichnisse müssen die Rechte
drwxrws---haben
Das „sticky“-Bit auf der Gruppenebene sorgt dafür, dass neu angelegte Dateien in diesem Vz. automatisch die Gruppenzugehörigkeit des Elternverzeichnisses erben (und nicht die des anlegenden Benutzers)
Falsch: drwx------ 35 tuid tuid 8192 Jun 17 23:19 /work/projects/…/myDir
Richtig: drwxrws--- 35 tuid <PrjID> 8192 Jun 17 23:19 /work/projects/…/myDir
Lösung:
Wechseln Sie in das Elternverzeichnis, unterhalb dessen das Problem-Verzeichnis liegt, und prüfen Sie dessen Zugriffsrechte mittels „ls -ld <Problemverzeichnis>“.. Stimmen sie nicht und gehört das Verzeichnis Ihnen statt der UNIX-Gruppe:
chgrp -R <PrjID> myDir
chmod 3770 myDir
Sollte das problematische Verzeichnis nicht Ihnen gehören, bitten Sie den Inhaber, diese Kommandos auszuführen.
Wir überarbeiten diese Seiten von Zeit zu Zeit.
Bitte senden Sie uns Ihre Frage oder Ihren VOrschlag als EMail an hhlr@hrz.tu-…. Dann können wir sie beantworten und gegebenenfalls hier aufnehmen.