
Verwechseln Sie nicht die Login-Knoten mit dem gesamten HPC-Cluster!
Zur Nutzung des Clusters reicht es nicht aus, ein Programm nur einfach auf dem Login-Knoten zu starten.
Die Loginknoten sind nicht für „produktive“ bzw. lang andauernde Rechnungen gedacht!

Mit der aktuellen Cluster-Konfiguration müssen im Regelfall keine Partitionen (oder Queues) beim Abschicken eines neuen Jobs angegeben werden – das erledigt Slurm weitestgehend automatisch anhand allgemeiner Job-Eigenschaften (z.B. max. Laufzeit und spezieller Ressourcen-Anforderung wie Akzeleratoren).
Nutzer mit mehreren Projekten müssen darauf achten, dass beim Abschicken neuer Rechenjobs die Projektzugehörigkeit korrekt spezifiziert wurde (z.B. mittels des Parameters -A <Projekt-Name>).
sbatch <Job-Script>
Stellt einen neuen Job in eine Queue (Warteschlange) ein. Die Optionen und Parameter für Kommandos und Job-Scripts findet man im folgenden Abschnitt Parameter des Kommandos „sbatch“.
Verschiedene kommentierte Beispiele für Job-Scripts sind ebenfalls unten auf dieser Seite zu finden..squeue
Zeigt den Stand und die Job-IDs aller Ihrer wartenden und aktiven Jobs.csqueue
Zeigt den Stand, die Job-IDs und andere grundlegende Informationen bezüglich aller wartenden und aktiven Jobs im Cluster.sjobs <Job-ID>
Das ist ein spezielles TU Darmstadt Script und zeigt detaillierte Informationen über alle Jobs bzw. über den Job (Job-ID) an.scancel <Job-ID<
Löscht einen Job aus der Warteschlange oder beendet einen aktiven Job.scancel -u $USER
Löscht/beendet alle eigenen Jobs.
csreport
Das ist ein spezielles TU Darmstadt Script und zeigt für alle Ihre Projekte den Gesamtverbrauch der vergangenen Monate an. Die Werte sind in Core*Stunden pro Monat angegeben, und dementsprechend prozentual auf das monatliche Budget bezogen (nicht auf das Gesamtbudget des Projekts).
Für den aktuellen Monat wird zusätzlich auch Ihr eigener Nutzeranteil gezeigt (wichtig bei Projekten mit mehreren Nutzern).sreport
Das ist das Slurm-Standardkommando zum Anzeigen des Ressourcen-Verbrauchs zu jedem ihrer Projekte.
Achtung: ohne „-t hours“ erfolgt die Ausgabe in Core*Minuten.- Mit diesem Standardkommando kann man auch selektiv für einen bestimmten Zeitraum die Werte anzeigen.
Dazu müssen die Parameter „cluster account“ (verbatim) sowie der Start- und Ende-Zeitpunkt angegeben werden.
Nachfolgendes Beispiel zeigt die Core-Hours für den Monat April 2024:sreport cluster Account Start=2024-04-01 End=2024-05-01 -t hours
- Mit diesem Standardkommando kann man auch selektiv für einen bestimmten Zeitraum die Werte anzeigen.
csum
Das ist ein spezielles TU Darmstadt Script und zeigt für alle ihre Projekte den aktuellen Gesamtverbrauch (im Vergleich zu den genehmigten Ressourcen) an Core*Stunden an.
Zur besseren Nachvollziehbarkeit empfehlen wir, alle Parameter explizit im Job-Script via „#SBATCH …“-Pragma aufzuführen, anstatt sie sbatch jedesmal als Kommandozeilenparameter zu übergeben. Auf diese Weise ist auch viel später noch nachvollziehbar, mit welchen Einstellungen ein Job gelaufen ist.
Beispiele für Job-Scripts (MPI, OpenMP, MPI+OpenMP=hybrid, GPU) finden Sie weiter unten auf dieser Seite.
Hier sind nur die wichtigsten Pragmas des sbatch-Kommandos angeführt. Eine vollständige Parameterliste liefert das Kommando ''man sbatch'' auf den Login-Knoten.
Der Batch-Scheduler benötigt mindestens die drei Parameter „--ntasks“, „--mem-per-cpu“ und „--time“, um entlang dieser Dimensionen Jobs auf die Rechenknoten zu verteilen.
-A Projekt-Bezeichnung
Für das Accounting kann man hiermit eins der eigenen Projekte auswählen, auf das die verwendeten Core-Stunden gebucht/abgerechnet werden.
Achtung: Jeder Nutzer hat ein default-Projekt (meist sein erstes angemeldetes), auf das automatisch gebucht wird, wenn man diese Option nicht angibt!
-J Job_Name
Weist dem Job einen frei wählbaren, beschreibenden Namen zu.
Referenzierbar als %x (in #SBATCH … pragmas) und als $SLURM_JOB_NAME (im Rest des Scripts).
--mail-type=BEGIN
Sendet eine E-Mail beim Start des Jobs.--mail-type=END
Sendet eine E-Mail beim Ende des Jobs.--mail-type=ALL
Sendet Mails bei beiden Vorgängen (und in einigen weiteren Fällen).
Bitte beachten: wenn man sehr viele Jobs einzeln abschickt, werden dementsprechend sehr viele Mails erzeugt. Das hat in der Vergangenheit dazu geführt, dass diverse eMail- bzw. Internet Service Provider die Mailserver der TU als „spammer“ auf ihre Blacklisten gesetzt haben und jegliche weitere Mailannahme verweigern.
Das Mail- und Groupware-Team des HRZ hatte dann sehr viel Mühe, dieses Blacklisting rückgängig zu machen.
Vermeiden Sie dies bitte mittels
- job arrays (
#SBATCH -a 1-100für 100 gleichartige Jobs) --mail-type=NONE– stattdessen mittels „squeue“ prüfen, welche Jobs noch laufen und welche fertig sind
Bitte vermeiden Sie auch, Ihre Mailadresse mittels „--mail-user=“ selbst zu spezifizieren – die leider nicht seltenen Tippfehler bei diesem Parameter kosten uns unnötigen Arbeitsaufwand. Unser Setup ermittelt auch ohne diese Angabe automatisch Ihre Mailadresse aus Ihrer TU-ID.
-o /pfad/zu/AusgabeDatei_Name (besser mit vollständigem Pfad)
Schreibt die Standard-Ausgabe STDOUT des gesamten Jobs-Scripts in die genannte Datei.-e /pfad/zu/FehlerDatei_Name (besser mit vollständigem Pfad)
Schreibt den Fehler-Kanal STDERR in die genannte Datei.
Bei beiden Optionen empfehlen wir, den vollen Pfadnamen und/oder „%.“-Dateinamen-Variablen anzugeben, um versehentliches Überschreiben anderer Job-Ausgaben und -Fehlerdateien zu vermeiden.
-n/--ntasks Task_Zahl
Gibt die Anzahl an (MPI-) Tasks (= separate Prozesse) für diesen Job an. Sofern nicht anders spezifiziert, stimmt dies mit der Gesamtanzahl der benötigten CPU-Rechenkerne überein, die der Job nutzen soll.
Prozesse kann der Scheduler immer auf verschiedene Rechenknoten verteilen.
(Dafür muss Ihr Programm MPI-fähig sein.)
MPI-Programme müssen unter der Kontrolle von „mpirun“ oder „srun“ (bevorzugt) gestartet werden, also z.B.
srun /path/to/my/MPIprogram …-N/--nodes Knoten_Zahl
Hiermit können Sie beeinflussen, auf wieviele verschiedene Rechenknoten Ihr MPI-Programm verteilt werden soll.
Fordern Sie z.B. „-n 384“ an, kann „-N 4“ helfen, die Prozesse dicht gepackt auf nur 4 Rechenknoten à 96 CPU-Rechenkerne zu verteilen, um ihren Interconnect-Verkehr zu minimieren.
-c Anzahl_Kerne_pro_Prozess (Voreinstellung: 1)
Gibt die Anzahl der Rechenkerne pro Task/Prozess an.
Für reine MultiThreading/OpenMP-Programme sollte man -n auf 1 setzen und -c auf die Anzahl der OpenMP-Threads.
Threads werden nie auf verschiedene Rechenknoten verteilt.
Programme, die nur per MultiThreading/OpenMP parallelisiert sind, dürfen nicht mittels „srun“ bzw. „mpirun“ gestartet werden!
Wenn Sie Ihr Programm immer exakt soviele Threads/Cores nutzen lassen wollen wie oben mit „#SBATCH -c …“ angefordert, fügen Sie die Zeile
export OMP_NUM_THREADS=$SLURM_CPUS_PER_TASK
in Ihr Jobscript (vor dem Programmstart) ein.
Auch alle anderen Angaben, wieviele CPU-Kerne/Threads ein Programm/Modul/Library nutzen soll (z.B. $MKL_NUM_THREADS), sollten besser mit dieser Variable gesetzt werden.
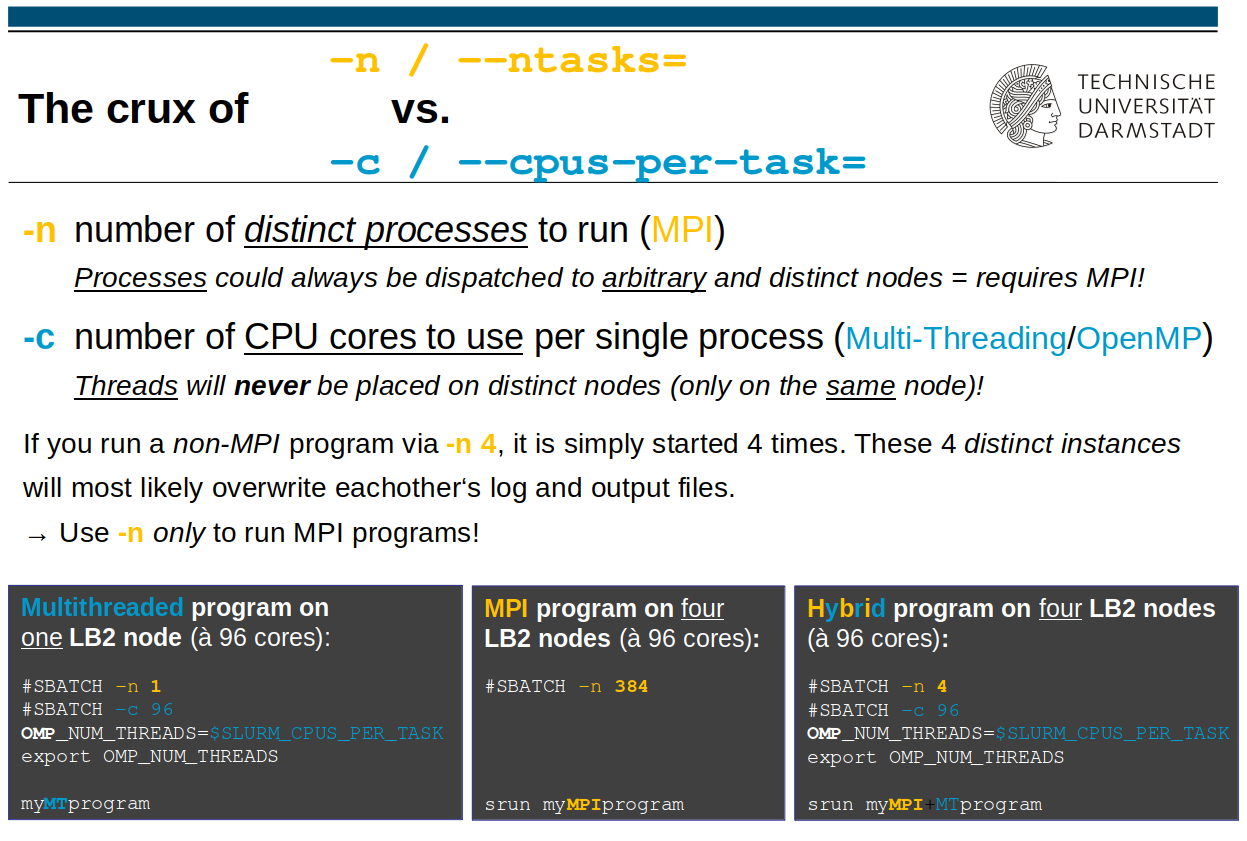
… liegt vor, wenn das Programm sowohl per MPI als auch mittels OpenMP/Multithreading parallelisierbar ist – zur Parametrisierung solcher Fälle siehe nebenstehendes Bild (rechter Fall).
Die Gesamtanzahl angeforderter CPU-Rechenkerne ergibt sich dann aus dem Produkt der mit „-n“ und „-c“ angegebenen Zahlen, also „Prozesse * Threads“.
In der Dokumentation Ihres Programms sollten Sie Hinweise finden, wann (bzw. für welchen seiner Algorithmen/Solver) welche Form der parallelen Ausführung am günstigsten ist, und mittels welcher Parameter Sie dem Programm diese Form vorgeben.
--mem-per-cpu=Speicher
Definiert den maximalen Hauptspeicher-Bedarf pro angefordertem Rechenkern in MByte. Hinweise zur Ermittlung dieses Werts für Ihr Programm finden Sie in den FAQs zum Batchsystem unter „Wie bestimme ich die Laufzeit-Parameter meines Jobs?“. Als problemloser Standard kann auf LB2 der Wert 2048 (2G) verwendet werden.
-t Laufzeit
Setzt das Laufzeit-Limit für den Job. Ein Job, der sich innerhalb dieser Zeit nicht selbst beendet, wird vom Batchsystem abgebrochen.
Die Zeit kann als einfache Zahl (= Minuten) oder detailliert in Form dd-hh:mm:ss (Tage-Stunden:Minuten:Sekunden) spezifiziert werden. Die übliche Laufzeit von 24h kann so als „-t 1-“ (ein Tag) abgekürzt werden.
-C feature
Fordert spezielle Features (Eigenschaften) für die Rechenknoten an, z.B. AVX512 oder größeren Hauptspeicher. Features dürfen mittels „&“ verknüpft werden.
Erlaubte Features sind u.a.:
avx512i01⇝ Rechenknoten der Ausbaustufe 1 oderi02⇝ Rechenknoten der Ausbaustufe 2memodermem1536gmpi(Standard)
-d job-dependencies
Hiermit lassen sich Abhängigkeiten zwischen verschiedenen Jobs definieren. Details sind unter ''man sbatch'' nachzulesen.
--exclusive
Fordert die Rechen-Knoten job-exklusiv an. Diese Option sollte für normale Batchjobs vermieden werden – kann aber wichtig sein, wenn man weniger Rechenkerne pro Knoten anfordert als vorhanden sind.
In so einem Fall (und ohne --exclusive) könnte Slurm weitere Jobs auf denselben Knoten schicken. Da dies das Laufzeitverhalten des ersten Jobs beeinflussen (und so möglicherweise Zeit- oder Performancemessungen verfälschen) könnte, stellt diese Option sicher, dass der damit gestartete Job allein auf dem Knoten ist.
--gres=Klasse:Typ:#: Beschleuniger-Anforderung, z.B. GPUs
(wenn nicht angegeben: Typ=<irgendeine Nvidia> und #=1)
--gres=gpu
fordert 1 Nvidia-GPU beliebigen Typs an--gres=gpu:3
fordert 3 Nvidia-GPUs beliebigen Typs an--gres=gpu:v100
fordert 1 Nvidia „Volta 100“-Karte an--gres=gpu:a100:3
fordert 3 Nvidia „Ampere 100“-Karten an--gres=gpu:h100:4
fordert 4 Nvidia „Hopper 100“-Karten an--gres=gpu:mi300x:6oder--gres=gpu:amd:6
fordert 6 AMD MI300X Karten an
(Infos zur Nutzung der MI300X (wird in neuem Tab geöffnet))--gres=gpu:pvc128g:2oder--gres=gpu:pvc:2
fordert 2 Intel „Ponte Vecchio“-Karten an
(Infos zur Nutzung der PVC (wird in neuem Tab geöffnet))
Um Ihre Job-Scripts nicht immer für wechselnde Anzahlen an GPUs doppelt umschreiben zu müssen, können Sie überall dort, wo Ihre Programme die Zahl der zu nutzenden GPUs erwarten, die Variable $SLURM_GPUS_ON_NODE verwenden.
Beispiel: „myCUDAprogram --num-devices=$SLURM_GPUS_ON_NODE“.
Da auf diese Weise immer ganze GPUs („Grafikkarten“) zugewiesen werden, gibt es keinen Weg, nur eine gewisse Anzahl an Tensor-Kernen oder Shadern oder G-RAM (Grafikkartenspeicher) pro GPU anzufordern. Wenn Ihrem Job eine GPU (Karte) zugewiesen wurde, können Sie daher alle ihre Ressourcen voll ausnutzen.
Wenn Sie für verteiltes Machine/Deep Learning mehrere GPU-Nodes benötigen (z.B. mit „horovod“), müssen Sie mit -N # (und dann -n >=#) explizit mehrere Nodes anfordern (wobei # = 2-8 gilt).
Da „GRes“ immer pro Node gelten, darf --gres=gpu:4 nur im Fall der DGX (8 GPUs) oder des AMD 300MIX Knotens überschritten werden, selbst wenn mehrere 4-GPU-Knoten angefordert werden.
MPI-Script
#!/bin/bash
#SBATCH -J <Job_Name>
#SBATCH --mail-type=END
#SBATCH -e /work/scratch/<TU-ID>/<yourWorkingDirectory>/%x.err.%j
#SBATCH -o /work/scratch/<TU-ID>/<yourWorkingDirectory>/%x.out.%j
#
#SBATCH -n 192 # Anzahl der Prozesse (= zu nutzende Rechenkerne, hier 2 Nodes à 96 Kerne)
#SBATCH --mem-per-cpu=2G # Hauptspeicher in MByte pro MPI-Task
#SBATCH -t 01:30:00 # in Stunden, Minuten und Sekunden, oder '#SBATCH -t 10' - nur Minuten
# -------------------------------
# Anschließend schreiben Sie Ihre eigenen Befehle zum Start der Berechnung, wie z.B.
module purge
module load gcc openmpi
cd /work/scratch/<TU-ID>/<yourWorkingDirectory>
srun <MPI-Programm> <Parameter>
EXITCODE=$?
# Aufräum- und Kopierbefehle:
...
# JobScript mit dem Status des wiss. Programms beenden
exit $EXITCODEMulti-Threading/OpenMP-Script
#!/bin/bash
#SBATCH -J <job_name>
#SBATCH --mail-type=END
#SBATCH -e /work/scratch/<TU-ID>/<project_name>/%x.err.%j
#SBATCH -o /work/scratch/<TU-ID>/<project_name>/%x.out.%j
#
#SBATCH -n 1 # nur 1 Prozess
#SBATCH -c 24 # Anzahl der Prozessorkerne pro Prozess
# kann unten als $SLURM_CPUS_PER_TASK abgerufen werden
#SBATCH --mem-per-cpu=2G # Hauptspeicher in MByte pro Prozessorkern
#SBATCH -t 01:30:00 # in Stunden, Minuten und Sekunden, oder '#SBATCH -t 10' - nur Minuten
# -------------------------------
# Anschließend schreiben Sie Ihre eigenen Befehle zum Start der Berechnung, wie z.B.
module purge
module load gcc
cd /scratch/<TU-ID>/<yourWorkingDirectory>
# falls Ihr Programm $OMP_NUM_THREADS beachtet/berücksichtigt:
export OMP_NUM_THREADS=$SLURM_CPUS_PER_TASK
</path/to/programm> <Parameter>
EXITCODE=$?
# Aufräum- und Kopierbefehle:
...
# JobScript mit dem Status des wiss. Programms beenden
exit $EXITCODEHybrid: MPI + OpenMP-Script
#!/bin/bash
#SBATCH -J <Job_Name>
#SBATCH --mail-type=END
#SBATCH -e /work/scratch/<TU-ID>/<Projekt_Name>/%x.err.%j
#SBATCH -o /work/scratch/<TU-ID>/<Projekt_Name>/%x.out.%j
#
#SBATCH -N 4 # Anzahl der Rechenknoten, auf die die -n Prozesse verteilt werden sollen
#SBATCH -n 4 # Anzahl der MPI-Prozesse/Tasks (vier Prozesse, aber - siehe nächste Zeile - jeder mit 96 Kernen)
#SBATCH -c 96 # Anzahl der Rechenkerne (OpenMP-Threads) pro MPI-Prozess
# ist unten als $SLURM_CPUS_PER_TASK abrufbar
#SBATCH --mem-per-cpu=2G # Hauptspeicher pro Rechenkern in MByte
#SBATCH -t 01:30:00 # in Stunden, Minuten und Sekunden, oder '#SBATCH -t 10' - nur Minuten
# -------------------------------
# Anschließend schreiben Sie Ihre eigenen Befehle zum Start der Berechnung, wie z.B.
module purge
module load gcc openmpi
cd /work/scratch/<TU-ID>/<yourWorkingDirectory>
# falls Ihr Programm $OMP_NUM_THREADS beachtet/berücksichtigt:
export OMP_NUM_THREADS=$SLURM_CPUS_PER_TASK
srun <Programm> <Parameter>
EXITCODE=$?
# Aufräum- und Kopierbefehle:
...
# JobScript mit dem Status des wiss. Programms beenden
exit $EXITCODEGPU/GRes
#!/bin/bash
#SBATCH -J <Job_Name>
#SBATCH --mail-type=END
#SBATCH -e /work/scratch/<TU-ID>/<Projekt_Name>/%x.err.%j
#SBATCH -o /work/scratch/<TU-ID>/<Projekt_Name>/%x.out.%j
#
# CPU-Anforderung
#SBATCH -n 1 # nur 1 MPI-Prozess/Task
#SBATCH -c 24 # Anzahl der Rechenkerne (OpenMP-Threads) pro MPI-Prozess
# ist unten als $SLURM_CPUS_PER_TASK abrufbar
#SBATCH --mem-per-cpu=2G # Hauptspeicher in MByte pro Rechenkern
#SBATCH -t 01:30:00 # in Stunden, Minuten und Sekunden, oder '#SBATCH -t 10' - nur Minuten
# GPU-Anforderung
#SBATCH --gres=gpu:v100:2 # 2 GPUs des Typs NVidia "Volta 100"
# ist unten als $SLURM_GPUS_ON_NODE abrufbar
# -------------------------------
# Anschließend schreiben Sie Ihre eigenen Befehle zum Start der Berechnung, wie z.B.
module purge
module load gcc cuda
cd /work/scratch/<TU-ID>/<yourWorkingDirectory>
# falls Ihr Programm $OMP_NUM_THREADS beachtet/berücksichtigt:
export OMP_NUM_THREADS=$SLURM_CPUS_PER_TASK
# nur zur Prüfung, ob und welche GPUs tatsächlich zugeteilt wurden
# (Ausgabe erscheint in der oben als "#SBATCH -e" definierten Datei):
nvidia-smi 1>&2
# falls Ihr Programm $CUDA_NUM_DEVICES beachtet/berücksichtigt:
export CUDA_NUM_DEVICES=$SLURM_GPUS_ON_NODE
./<Programm> -<Parameter>
EXITCODE=$?
# Aufräum- und Kopierbefehle:
...
# JobScript mit dem Status des wiss. Programms beenden
exit $EXITCODEDie Angabe „--gres=Klasse:Typ:Anzahl“ bezieht sich immer auf einen einzelnen Beschleuniger-Rechenknoten, und auf ganze GPU-Karten. Es gibt keinen Weg, explizit nur eine gewisse Anzahl an GPU-Kernen (z.B. 48 Tensor-Units) einer GPU anzufordern – man kann nur (mehrere) ganze GPU-Karten anfordern.
Interaktives Arbeiten auf Rechenknoten ist prinzipiell möglich, allerdings ist es insbesondere bei einer hohen Clusterauslastung sehr unpraktisch. Außer zum Debugging ist es immer besser, nicht-interaktive Jobs im Batch-System zu verwenden.
Falls interaktives Arbeiten unbedingt notwendig ist, kann dies mit dem srun Kommando und den Parametern --pty /bin/bash angefordert werden. Die drei immer notwendigen Parameter -t (Zeit), -n (Task Zahl) und --mem-per-cpu= (Speicher pro CPU-Kern) müssen ebenfalls angegeben werden. Optionale Parameter wie z.B. features und mail Optionen können ebenfalls auf der Kommandozeile angegeben werden.
Beispiel:
srun -t15 -n1 -c24 -C i01 --mem-per-cpu=500 --pty /bin/bashwürde einen Prozess (auf einem LB2A1-Knoten) mit 24 Rechenkernen für eine Viertelstunde anfordern.
Sowohl sbatch als auch srun kennen den Parameter „--test“, um auf
- syntaktische Korrektheit,
- das Vorhandensein aller erforderlichen Parameter und
- das generelle Vorhandensein der angeforderten Ressourcen-Konfiguration
zu testen, ohne wirklich einen Job abzuschicken („Probelauf“).
Generell ist der direkte Login auf die Rechenknoten nicht möglich.
Die einzige Ausnahme sind Knoten, auf denen gerade einer Ihrer Jobs läuft – vom Loginknoten aus (nicht aus dem Internet) können Sie sich dann auf diese Knoten weiterverbinden.
Das dient dazu, Ihrem Job detaillierter bei der Arbeit „über die Schulter zu schauen“ bzw. zu beurteilen, wie (und ob) er die zugewiesenen Ressourcen auch gut ausnutzt. Dazu können Sie Kommandos wie top, htop, oder auch strace oder andere Debugger bzw. Ihre eigenen Linux-Werkzeuge nutzen.
Da die Rechenknoten nicht direkt mit dem Internet verbunden sind, funktioniert das nur von den Loginknoten aus.
Die (Liste der) ausführenden Rechenknoten erhalten Sie vom Kommando
squeue -t RUNNINGin der Spalte NODELIST.
Im Fall von MPI-Jobs, die auf mehreren Knoten laufen, müssen Sie die kompakte Darstellung der NODELIST zuerst in einzelne Hostnamen zerlegen – beispielsweise ergibt „mpsc0[301,307,412-413]“ die Rechenknoten
mpsc0301
mpsc0307
mpsc0412
mpsc0413 Melden Sie sich dann (vom Loginknoten aus) auf einem von diesen an:
ssh <Hostname aus NODELIST>oder per
srun --overlap --jobid=<IhreJobID> --pty /bin/bash Wollen Sie auch damit einen bestimmten Rechenknoten ansteuern, fügen Sie vor „--pty“ noch dessen Hostnamen mit „-w mpsc…“ ein.
Job Arrays
Leider kann man „srun --jobid …“ nicht direkt eine Job Array-Task-ID wie „45678901_107“ übergeben. Rufen Sie „scontrol show job 45678901_107 | grep JobId=“ auf, um dessen interne JobID zu erfahren, und setzen Sie diese bei „srun --jobid …“ ein.
Ende des Jobs
Sollten Sie noch eingeloggt sein, wenn Ihr Job endet, wird auch Ihre separate ssh- bzw. srun-Sitzung zum Rechenknoten beendet, und Sie sind wieder auf dem Loginknoten.