
Do not confuse the login nodes with the HPC cluster!
To use the cluster, it is not sufficient to simply start your program on a login node.
The login nodes are not intended for “productive” or long-running calculations!

With the current cluster configuration, you normally do not need to specify a queue or partition when submitting new compute jobs, as this will be done automatically by Slurm, depending on the job's properties (e.g. the run time or special resources like accelerators).
Members of multiple projects however, should make sure to choose the proper project when submitting new jobs (e.g. with the parameter -A <project name>).
sbatch <job script>
This puts a new job in a queue.
Please refer to sbatch parameters for switches and options of commands and in job scripts.
Commented examples of job scripts are shown down below, too.squeue
This shows an overview of all your active and waiting jobs of the job queue.csqueue
This shows a global overview of all the active and waiting jobs of job queue.sjobs<Job-ID>
This is a special TU Darmstadt script for showing detailed information about all your pending and running jobs or the job with the given ID.scancel <Job-ID>
This deletes a job from the queue or terminates an active job.scancel -u $USER
Deletes/terminates all your own jobs.
csreport
This is a special TU Darmstadt script for showing the resource usage for each of your projects in the last months. All values are shown in core*hours per month, and are thus percent-related to your monthly share (not to your project's total budget).- For the current month you also see your own part of usage for each of your projects (important for projects with multiple users).
sreport
This is the standard Slurm command and shows the resource usage separated for each of your projects.
Attention: without “-t hours”, the values are given in core*minutes.- In addition you can get a report of a specific month or any time period. For that you need to give the parameters “
cluster account” (verbatim) and the start and end date.
The following example shows how to get a core*h report of April 2024:sreport cluster account Start=2024-04-01 End=2024-05-01 -t hours
- In addition you can get a report of a specific month or any time period. For that you need to give the parameters “
csum
This is a special TU Darmstadt script for showing the resource usage in total for each of your projects (in comparison to the approved value). This command shows all values in core*hours.
To enhance reproducibility, we recommend providing all parameters inside the job script as “#SBATCH …” pragma (instead of using sbatch command line parameters). This way, even rather old job scripts clearly document the conditions the job ran under.
Examples of different job scripts (MPI, OpenMP, MPI+OpenMP=hybrid, GPU) can be found down below.
Here, only the most important pragmas are given. You can find a complete list of parameters using the command ''man sbatch'' on the login nodes.
The batch scheduler requires at least the three parameter “--ntasks”, “--mem-per-cpu” and “--time”, to plan and distribute jobs along these dimensions onto compute nodes.
-A project_name
With this option, you choose the project the core hours used will be accounted on.
Attention: If you omit this pragma, the core hours used will be accounted on your default project (typically your first or main project), which may or may not be intended!
-J job_name
This gives the job a more descriptive name.
Referable as %x (in #SBATCH … pragmas) and as $SLURM_JOB_NAME (in the job script's payload).
--mail-type=BEGIN
Send an email at begin of job.--mail-type=END
Send an email at end or termination of job.--mail-type=ALL
Send an email at both events (and in some other special cases).
Please note: if you submit a lot of distinct jobs separately, at least the same number of emails will be generated. In the past, this problem has caused the mail servers of the TU Darmstadt to be blacklisted as “spamming hosts” by several mail and internet service providers, refusing to receive any further mail from the TU Darmstadt.
The mail and groupware team of the HRZ was having a lot of efforts to revert this.
Avoid this by
- using job arrays (
#SBATCH -a 1-100for 100 similar jobs) --mail-type=NONE– instead, use “squeue” to see all your jobs still running or finished.
Please also avoid setting “--mail-user=<yourMailAddress>”, as our system determines your mail address automatically based on your TU-ID, and typos in this parameter cause unnecessary workload.
-o /path/to/outfile_name
This writes the standard output (STDOUT) of the whole job script in the designated file.-e /path/to/errfile_name
This writes the error channel (STDERR) of the whole job script in the designated file.
For both options, we recommend to use the full pathname and/or to use %. file name variables, to avoid overwriting other job's files.
-n/--ntasks number of tasks
This determines the number of (MPI) tasks (separate processes) for this job. For MPI programs, this usually corresponds to the total number of necessary compute cores for the MPI job.
Processes can always be scheduled to different nodes
(for that, your program is required to be capable of using MPI).
Also, MPI programs need to be run under control of either “mpirun” or “srun” (preferred), ie.
srun /path/to/my/MPIprogram …-N/--nodes #_nodes
This helps to control on how many distinct compute nodes your MPI programm will be distributed..
If you request for example “-n 384” CPU cores, specifying “-N 4” can help in densely packing your MPI processes to only 4 compute nodes à 96 CPU cores, reducing their amount of interconnect traffic.
-c cores_per_task (Default: 1)
This gives the number of cores per task/process. For pure multi-threading/OpenMP jobs, -n should be set to 1 and -c to the number of OpenMP threads.
Threads will never be scheduled onto distinct nodes.
Pure multi-threaded/OpenMP programs must not be started under “srun” or “mpirun”.
If you always want your OpenMP program to use the allotted number of cores requested with “#SBATCH -c …”, use the following construct in your job script (before the program's line):
export OMP_NUM_THREADS=$SLURM_CPUS_PER_TASK.
Any other directives for how many cores/threads your program/module/library should use (eg. $MKL_NUM_THREADS) are best specified using this $SLURM_CPUS_PER_TASK variable, too.
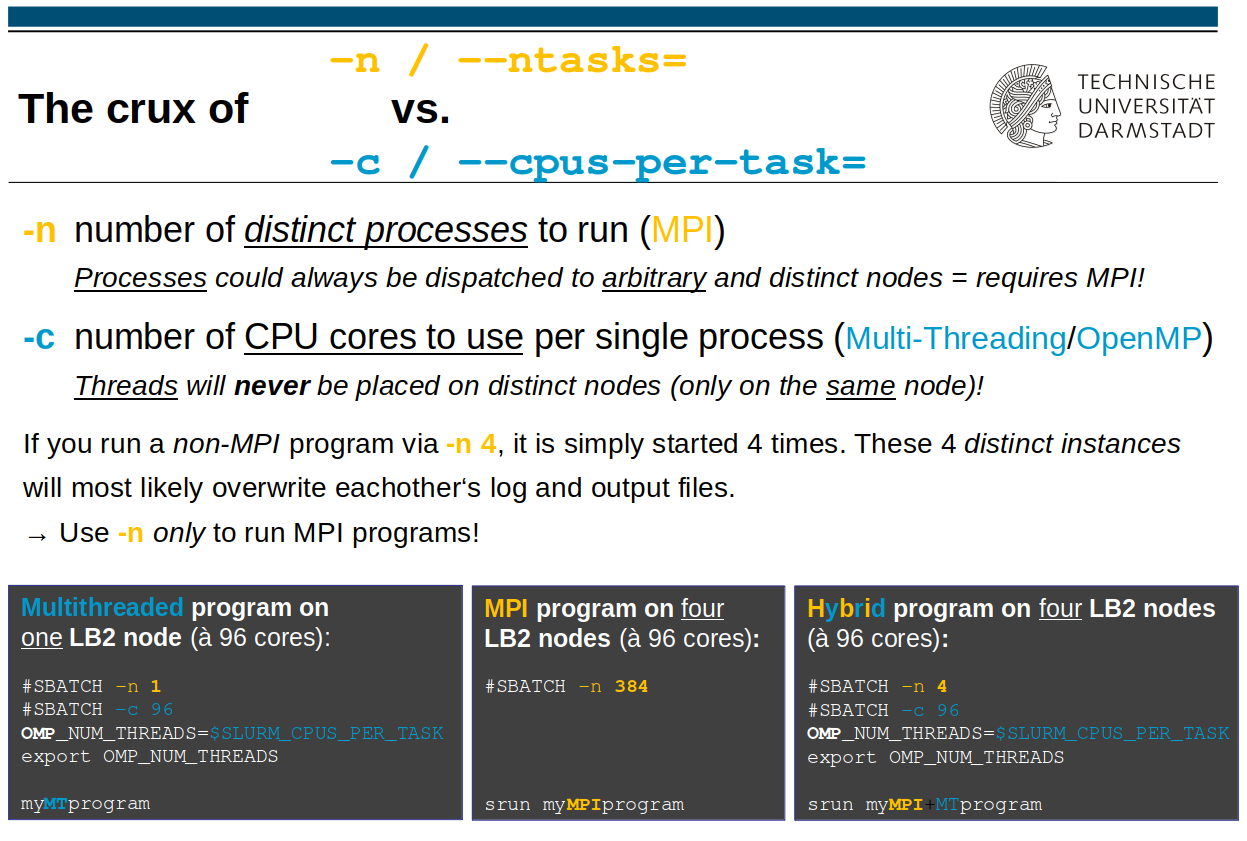
… is when your program can use MPI and OpenMP/Multithreading for parallelization – see the adjacent picture (rightmost case) for how to handle that).
Then, the total amount of CPU cores requested is the multiplicate of the “-n” und “-c” values, i.e. “processes * threads”.
The documentation of your program should give you clues as to when (or for which of its algorithms/solvers) to use which form of parallelization, and how to parameterize your program to do so.
--mem-per-cpu=memory
This defines the maximum required main memory per compute core in MByte.
For how to get an idea of this value for your program, see the FAQ batch system, heading How Do I “Size” My Job?. If you are uncertain, you can start with a default of 2048 (2G) on LB2.
--time runtime
This sets the run time limit for the job (“wall clock time”). If a job is not completed within this time, it will be terminated by the batch system.
The prospected run time can be given as simple number (= minutes) or specified en detail as dd-hh:mm:ss (days-hours:minutes:seconds). The usual 24h runtime can thus be abbreviated as “-t 1-” (one day).
-C feature
Requests nodes for this job to have a certain feature, e.g. AVX512 or larger main memory. Features can be combined by “&”.
Valid features are for example:
-d dependency
This determines dependencies between different jobs. For details, please see ''man sbatch''.
--exclusive
This requests a compute node job-exclusively, meaning there are no other jobs allowed on this node.
Usually unnecessary for normal jobs. This might only be important if you request fewer cores than available per node. In this case (and in absence of --exclusive), Slurm could dispatch other jobs to the node. As this could adversely affect the runtime behaviour of the first job (possibly distorting timing and performance analyses), this option makes sure the job is the only one on its node.
--gres=class:type:# accelerator specification, eg. GPUs
(if not specified, the defaults are: type=<any Nvidia> and #=1)
--gres=gpu
requests 1 Nvidia GPU accelerator card--gres=gpu:3
requests 3 Nvidia GPU accelerator cards--gres=gpu:v100
requests 1 Nvidia “Volta 100” card--gres=gpu:a100:3
requests 3 Nvidia “Ampere 100” cards--gres=gpu:h100:4
requests 4 Nvidia “Hopper 100” cards--gres=gpu:mi300x:6or--gres=gpu:amd:6
requests 6 AMD MI300X GPUs
(MI300X usage info (opens in new tab))--gres=gpu:pvc128g:2or--gres=gpu:pvc:2
requests 2 Intel “Ponte Vecchio” GPUs
(PVC usage info (opens in new tab))
To have your job scripts (and programs) adapt automatically to the amount of (requested) GPUs, you can use the variable $SLURM_GPUS_ON_NODE wherever your programs expect the number of GPUs to use, ie.
“myCUDAprogram --num-devices=$SLURM_GPUS_ON_NODE”.
Since you always get whole GPU cards allocated, there is no way of asking for a certain amount of tensor cores or shaders or G-RAM (GPU memory) in a GPU. When allocated, you are free to use each and every resource inside that allocated GPU card at will.
If you need more than one GPU compute node for distributed Machine/Deep Learning (eg. using “horovod”), the job needs to request several GPU nodes explicitly using -N # (with # = 2-8). Consequently, the number of tasks requested with -n # needs to be equal or higher than the number of nodes.
Since “GRes” are per node, you should not exceed --gres=gpu:4 (except when targeting the DGX with 8 GPUsor the AMD MI300X GPUs), even when using several 4-GPU-nodes.
MPI-Script
#!/bin/bash
#SBATCH -J <Job_Name>
#SBATCH --mail-type=END
#SBATCH -e /work/scratch/<TU-ID>/<yourWorkingDirectory>/%x.err.%j
#SBATCH -o /work/scratch/<TU-ID>/<yourWorkingDirectory>/%x.out.%j
#
#SBATCH -n 192 # number of processes (= total cores to use, here: 2 nodes à 96 cores)
#SBATCH --mem-per-cpu=2G # required main memory in MByte per MPI task/process
#SBATCH -t 01:30:00 # in hours, minutes and seconds, or '#SBATCH -t 10' - just minutes
# -------------------------------
# your job's "payload" in form of commands to execute, eg.
module purge
module load gcc openmpi
cd /work/scratch/<TU-ID>/<yourWorkingDirectory>
srun <MPI program> <parameters>
EXITCODE=$?
# any cleanup and copy commands:
...
# end this job script with precisely the exit status of your scientific program above:
exit $EXITCODEMulti-Threading/OpenMP-Script
#!/bin/bash
#SBATCH -J <job_name>
#SBATCH --mail-type=END
#SBATCH -e /work/scratch/<TU-ID>/<project_name>/%x.err.%j
#SBATCH -o /work/scratch/<TU-ID>/<project_name>/%x.out.%j
#
#SBATCH -n 1 # 1 process only
#SBATCH -c 24 # number of CPU cores per process
# can be referenced as $SLURM_CPUS_PER_TASK in your "payload" down below
#SBATCH --mem-per-cpu=2G # Main memory in MByte for each cpu core
#SBATCH -t 01:30:00 # Hours and minutes, or '#SBATCH -t 10' - just minutes
# -------------------------------
# your job's "payload" in form of commands to execute, eg.
module purge
module load gcc
cd /work/scratch/<TU-ID>/<project_name>
# Specification from OMP_NUM_THREADS depends on your program
export OMP_NUM_THREADS=$SLURM_CPUS_PER_TASK
</path/to/program> <parameters>
EXITCODE=$?
# any cleanup and copy commands:
...
# end this job script with precisely the exit status of your scientific program above:
exit $EXITCODEHybrid: MPI + OpenMP-Script
#!/bin/bash
#SBATCH -J <Job_Name>
#SBATCH --mail-type=END
#SBATCH -e /work/scratch/<TU-ID>/<project_name>/%x.err.%j
#SBATCH -o /work/scratch/<TU-ID>/<project_name>/%x.out.%j
#
#SBATCH -N 4 # number of compute nodes to distribute the -n processes to
#SBATCH -n 4 # number of processes (here: just 4, but - see next line - each with 96 cores)
#SBATCH -c 96 # number of OpenMP threads or CPU cores per process
# can be referenced as $SLURM_CPUS_PER_TASK in your "payload" down below
#SBATCH --mem-per-cpu=2G # Main memory in MByte for each cpu core
#SBATCH -t 01:30:00 # Hours and minutes, or '#SBATCH -t 10' - just minutes
# -------------------------------
# your job's "payload" in form of commands to execute, eg.
module purge
module load gcc openmpi
cd /work/scratch/<TU-ID>/<yourWorkingDirectory>
# specification from OMP_NUM_THREADS depends on your program
export OMP_NUM_THREADS=$SLURM_CPUS_PER_TASK
srun <Programm> <Parameter>
EXITCODE=$?
# any cleanup and copy commands:
...
# end this job script with precisely the exit status of your scientific program above:
exit $EXITCODE
GPU/GRes
#!/bin/bash
#SBATCH -J <Job_Name>
#SBATCH --mail-type=END
#SBATCH -e /work/scratch/<TU-ID>/<project_name>/%x.err.%j
#SBATCH -o /work/scratch/<TU-ID>/<project_name>/%x.out.%j
#
# CPU specification
#SBATCH -n 1 # 1 process
#SBATCH -c 24 # 24 CPU cores per process
# can be referenced as $SLURM_CPUS_PER_TASK in the "payload" part
#SBATCH --mem-per-cpu=2G # Hauptspeicher in MByte pro Rechenkern
#SBATCH -t 01:30:00 # in hours:minutes, or '#SBATCH -t 10' - just minutes
# GPU specification
#SBATCH --gres=gpu:v100:2 # 2 GPUs of type NVidia "Volta 100"
# can be referenced down below as $SLURM_GPUS_ON_NODE
# -------------------------------
# your job's "payload" in form of commands to execute, eg.
module purge
module load gcc cuda
cd /work/scratch/<TU-ID>/<yourWorkingDirectory>
# specification from OMP_NUM_THREADS depends on your program
export OMP_NUM_THREADS=$SLURM_CPUS_PER_TASK
# for checking whether and which GPUs have been allocated
# (output appears in the "#SBATCH -e" file specified above):
nvidia-smi 1>&2
# if your program supports this way of getting told how many GPUs to use:
export CUDA_NUM_DEVICES=$SLURM_GPUS_ON_NODE
./<Programm> <Parameter>
EXITCODE=$?
# any cleanup and copy commands:
...
# end this job script with precisely the exit status of your scientific program above:
exit $EXITCODE
The request “--gres=Class:Type:Amount” always refers to a single accelerator node, and to GPU cards as a whole. There is no way of requesting separate amounts of GPU cores (i.e. 48 Tensor units)--you can just ask for (several) whole GPU cards.
In principle. it is possible to work interactively on compute nodes, though it is not advised for regular work: due to many pending jobs and a mostly fully used-up cluster, resources will not be available immediately.
If interactive work on compute nodes in fact is necessary, it can be requested with the srun command and the --pty /bin/bash options. The three mandatory parameters are -t (time), -n (No. of tasks) and --mem-per-cpu= (memory per task), and need to be supplied to the srun command, too. Optional parameters like features and mail options can also be given on the command line.
Example
srun -t15 -n1 -c24 -C i01 --mem-per-cpu=500 --pty /bin/bashwould request one process (on one LB2A1 node) with 24 CPU cores for 15 minutes.
Both sbatch as well as srun accept a “--test” parameter to check for
- syntactical correctness,
- presence of all mandatory parameters, and
- general existence of the requested hardware resources
all without really submitting a job (aka “dry run”).
In general, a direct login to the compute nodes is not possible.
However, during execution of your own job(s), you are entitled to login to the executing compute nodes (from a login node, not from the internet).
This allows you to have a detailed look on how (and if) your job is using the assigned resources. You can use commands like top, htop, debugging tools like strace or your own favorite linux tools, and in general to see the behaviour of your job(s) at first-hand.
This works only from the login node, as the compute nodes are not directly connected to the internet.
To get the (list of) compute node(s) executing your job, run
squeue -t RUNNINGand inspect the “NODELIST”.
In case of multi-node MPI jobs, the compact NODELIST needs to be decomposed into distinct host names. For example, the node list “mpsc0[301,307,412-413]” expands to
mpsc0301
mpsc0307
mpsc0412
mpsc0413which you are all entitled to log in to, while these are still executing your job.
Then hop onto one of them (from a login node only) by either executing
ssh <name of compute node (as decomposed) from NODELIST>or
srun --overlap --jobid=<yourJobID> --pty bashIf you here would like specify a certain one of them, insert its name (before the “--pty” parameter) as “-w mpsc…”.
If your job ends while you are still logged into one of its compute nodes, you will be logged out automatically, ending up back on the login node.